Keywords: Melanoma; MEK; BRAF; Trametinib; Trial; Phase 1.
Abstract
Background: The maximal tolerated dose of the MEK inhibitor trametinib with weekly paclitaxel was sought, with a view to exploring the combination’s activity in melanoma lacking a BRAF V600 mutation.
Methods: In this phase 1 study a fixed dose of paclitaxel (80 mg/m2 intravenous (IV) on days 1, 8, and 15 of each 4‑week cycle) was used and the dose of Trametinib GSK1120212 MEK inhibitor melanoma was escalated (to a maximum 2 mg orally (PO) daily), following a 3 + 3 design; eligible patients had advanced melanoma and could have received up to two previous lines of treatment for metastatic disease.
Findings: Fifteen patients were enrolled, all but one of whose melanoma was wild type for BRAF at codon 600; the maximal monotherapy dose of trametinib proved tolerable with weekly paclitaxel, the most frequent adverse events were rash and fatigue, six (40%) partial responses were reported including four of eight patients with NRAS mutations, median progression‑free survival was 5.5 months (95% confidence interval (CI) 1.8–7.8 months) and overall survival was 14.1 months (95% CI 4.6–not reached).
Interpretation: Trametinib can safely be given with weekly paclitaxel at the full monotherapy dose, and in this small group, promising progression‑free and overall survival were observed in patients with melanoma lacking a V600 BRAF mutation.
Introduction
Melanoma remains a deadly disease with a rising incidence, with almost 80,000 cases diagnosed and over 10,000 deaths reported in the United States of America in 2013, and despite recent treatment advances, median survival is still measured in months.
A greater understanding of the genetic landscape of melanoma has led to successful development of targeted treatments, with the mitogen‑activated protein (MAP) kinase pathway implicated in melanoma growth and frequently activated through pathological mutations in BRAF or NRAS.
Selective inhibition of mutant BRAF has resulted in significantly improved clinical outcomes, with objective response rates of approximately 50% and significantly improved progression‑free and overall survival compared with chemotherapy in V600 mutant BRAF melanoma, and combined inhibition at RAF and MEK is associated with even greater response rates and better progression‑free survival.
In contrast, patients with melanomas lacking an activating BRAF mutation have fewer therapeutic options currently limited to chemotherapy or immunotherapy with the cytotoxic T‑lymphocyte antigen 4 (CTLA‑4) antagonist ipilimumab, and although newer treatments targeting programmed death 1 (PD‑1) and its ligand have raised expectations, only a minority benefit and more effective treatments are needed particularly for BRAF V600 wild‑type melanoma.
Extracellular‑signal regulated kinase (ERK) is activated following upstream phosphorylation of MEK and RAF in the MAP‑kinase pathway, with melanoma cells showing high levels of constitutive ERK activation irrespective of mutation status; pathway activation is implicated in resistance to taxane chemotherapy via ERK1/2‑mediated degradation of Bim and phosphorylation of Bad, inhibiting apoptosis, and co‑administration of a MEK inhibitor and taxane induces apoptosis and tumour regression in animal xenograft models in both wild‑type and V600E mutated BRAF melanoma cell lines.
In clinical trials, combinations of taxanes and MEK inhibitors have shown early signs of efficacy, including a placebo‑controlled phase 2 study where selumetinib plus docetaxel improved progression‑free survival and overall response rate in K‑RAS mutant non‑small cell lung cancer compared to docetaxel alone, and a similar trial in BRAF wild‑type melanoma showed non‑significant increases in progression‑free survival and response rate.
Trametinib is a reversible, highly selective, allosteric inhibitor of MEK1/MEK2 activation with greater clinical activity in mutant BRAF melanoma than selumetinib; in the phase 3 METRIC study trametinib demonstrated superior progression‑free survival (median 4.5 versus 1.5 months; p < 0.001) and 6‑month survival (81% versus 67%; p = 0.01) compared to chemotherapy, whereas selumetinib had similar activity to temozolomide in a 200‑patient phase 2 study, and trametinib’s pharmacokinetic profile suggests sustained target inhibition throughout dosing, making it attractive for combination therapy.
Therefore, paclitaxel was combined with trametinib in a phase 1 trial involving patients with melanoma to establish a tolerable combination for a randomised phase 2 trial comparing this combination with paclitaxel alone in wild‑type BRAF melanoma.
Methods
2.1. Participants
Patients were enrolled from three institutions and were required to be 18 years or older with unresectable stage III or stage IV cutaneous melanoma, Eastern European Oncology Group (ECOG) scores of zero or one, and acceptable haematological, renal, and hepatic laboratory values.
Exclusion criteria included receiving more than two previous lines of therapy for advanced disease, prior treatment with a MEK inhibitor or taxane, recent systemic therapy or radiotherapy, recent history of another active malignancy, and ocular disease predisposing to central serous retinopathy or retinal vein occlusion, though patients with stable brain metastases were eligible, and all patients provided written consent.
2.2. Study design
This dose‑escalation study investigated the maximum tolerated dose (MTD) of trametinib in combination with a fixed weekly dose of paclitaxel (80 mg/m2 on days 1, 8, and 15 of each 4‑week cycle), with trametinib administered orally once daily at 1.0, 1.5, or 2.0 mg and permitted beyond six cycles of paclitaxel for patients continuing to benefit.
Cohorts of three patients were recruited at each dose level with planned expansion to six patients if a single dose‑limiting toxicity (DLT) occurred, treatment continued until disease progression or predefined unacceptable toxicity, adverse events were graded by CTCAE version 4.0 with weekly assessments during chemotherapy, DLTs were defined as drug‑related grade 3 non‑haematological events, grade 4 haematological events, persistent visual disturbance, febrile neutropenia in cycle one, or a delay of more than 2 weeks to start cycle two, and MTD was defined as the dose at which no more than two of six patients experienced a DLT.
Tumours were assessed radiologically every 8 weeks and then every 3 months following chemotherapy using RECIST 1.1, and the protocol was approved by review boards at all participating institutions.
2.3. Statistical analysis
Sample size was expected to be between six and eighteen patients according to observed toxicities, adverse events were summarised descriptively by dose level, progression‑free survival (PFS) was defined from registration to progression or death with censoring at latest assessment for those without events, overall survival (OS) was defined from registration to death with censoring at last visit for those without events, and objective response rate (ORR) was defined as the proportion achieving complete or partial response as best overall response.
2.4. Role of funding source
The study was conducted by the Oncology Clinical Trials Office (OCTO) and the University of Oxford, supported by an educational grant from GlaxoSmithKline under the NIHR Clinical Research Network (CRN) Cancer Division Industry Alliance Program.
Results
3.1. Baseline characteristics
Between April 2012 and February 2013, sixteen patients were enrolled, one of whom had imaging confirming a tumour response prior to the trial and did not proceed with treatment, leaving fifteen treated patients.
Most patients had not received prior systemic treatment, the majority had normal lactate dehydrogenase (LDH), one patient had a V600E BRAF mutation, eight patients (53%) had an NRAS mutation at codon 61, and tumour NRAS status was not assessed in one patient due to insufficient tissue.
3.2. Safety
The first patient in cohort one (1.0 mg daily trametinib) developed a DLT and the cohort was expanded from three to six patients, with no further DLTs in that cohort or in cohort two (1.5 mg daily).
The first three patients in cohort three (2.0 mg daily) completed cycle one uneventfully and the cohort was expanded to six to confirm safety at the maximal daily trametinib dose, with the fifth patient experiencing a DLT and the sixth requiring a trametinib dose delay during cycle one; as four of six tolerated the maximal dose, the MTD was determined at this dose level.
Patients received trametinib for a median of 111 days with 40% continuing for at least five months, and at the most recent data lock two patients were still receiving treatment for 456 and 391 days, respectively.
Four patients (26%) required trametinib dose reductions, paclitaxel administration was delayed by one week in two patients and dose reduced in one patient in cycle five.
Two DLTs were reported: central serous retinopathy in cohort one resolved after a 2‑week trametinib interruption, and grade 4 liver function test abnormalities in cohort three normalised within 2 weeks after trametinib cessation without recurrence despite ongoing paclitaxel.
Serious adverse events occurred at least once in eight patients (53%) and twice or more in four patients (26.6%), with infection most common in two patients (13.3%), and the most common adverse events were rash and fatigue.
Rash was predominantly acneiform, reversible, not dose‑dependent, responsive to topical treatments (corticosteroid‑based creams and topical antibiotics), and significant episodes (grade three or higher) resolved within a fortnight following trametinib interruption and remained manageable with dose reductions, with no cases of squamoproliferative lesions reported.
A single case of ocular hypertension resolved within 2 weeks of trametinib interruption and did not recur with dose reduction, and retinal vein occlusion was not reported.
Hepatotoxicity occurred in three patients (20%), with grade 2 cases not requiring modification and one grade 4 episode improving with trametinib cessation despite ongoing paclitaxel.
A possible treatment‑related fatality occurred in cohort one in a patient who developed grade 3 fatigue after four cycles; both drugs were withheld, but the patient deteriorated with a pulmonary embolism, worsening liver function tests, and oliguric renal failure, abdominal imaging showed hepatic steatosis, an upper gastrointestinal haemorrhage occurred during intensive therapy support, acute tubular necrosis was considered the primary cause of death though not histologically confirmed, imaging excluded progressive disease, and the death was regarded as possibly related to trametinib.
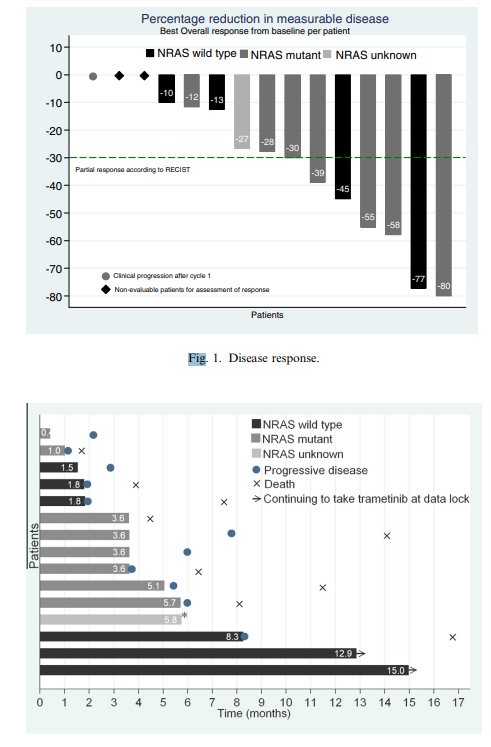
Figure 1. Disease response.
Figure 2. Total time receiving trametinib and time to progressive disease and death according to NRAS mutational status; treatment stopped (due to toxicity) in the absence of disease progression or death indicated by asterisk.
3.3. Efficacy
Of the fifteen treated patients, two did not have measurable disease at baseline and one progressed clinically during cycle one; among the remaining twelve patients, all had tumour reduction ranging from 10% to 77%, six patients (40%) experienced a partial response, four (27%) were confirmed on subsequent imaging, four unconfirmed partial responses occurred in NRAS‑mutant patients (half of those with this mutation), extent of tumour shrinkage did not appear to relate to trametinib dose level, median PFS for the entire cohort was 5.5 months (95% CI 1.8–7.8), and OS was 14.1 months (95% CI 4.6–not reached).
Median PFS and OS were unchanged when excluding the BRAF‑mutant patient, patients with NRAS mutations (n = 8) had median PFS of 4.6 months (95% CI 1.2–6.1) and OS of 8.1 months (95% CI 1.7–not reached), and two NRAS and BRAF wild‑type patients had PFS greater than 12 months and continued on trametinib at data lock.
Figure 3. Progression‑free (A) and overall survival (B) for the fourteen patients with BRAF V600E wild‑type melanoma (median PFS = 5.5 months (95% CI 2.3–8.1), median OS = 14.1 months (95% CI 6.4–not reached)).
Discussion
Patients with BRAF wild‑type metastatic melanoma have fewer options than those with mutant disease, with standard treatments outside clinical trials currently limited to immunotherapy and cytotoxic chemotherapy, where paclitaxel and dacarbazine produce responses in under 10% with most progressing within two months, and immune checkpoint inhibitors such as ipilimumab, nivolumab, and pembrolizumab offer durable responses for a minority.
This study provides evidence of activity in BRAF wild‑type melanoma, reporting six (40%) partial responses (four (26%) confirmed) with median PFS and OS of 5.5 and 14.1 months respectively, and although not designed to assess efficacy and limited by modest numbers, the data compare intriguingly to other trials in similar populations.
In a large phase 1 trial of trametinib monotherapy in advanced melanoma, among 39 BRAF wild‑type patients the response rate was 10% versus 40% in BRAF‑mutant patients and median PFS was 2.0 months (95% CI 1.7–3.7 months), while a phase 2 trial comparing temozolomide to selumetinib reported only one partial response in the BRAF wild‑type subpopulation with median PFS of 2.6 months for selumetinib similar to temozolomide.
A phase 2 placebo‑controlled trial of docetaxel with or without selumetinib in strictly BRAF wild‑type melanoma suggested increased activity for the combination but did not detect PFS or OS differences, attributed to use of an inferior MEK inhibitor, and PFS in the selumetinib arm (4.23 months) was less than the PFS observed here (5.5 months), which is comparable to vemurafenib’s PFS (5.3 months) in BRIM‑3 for BRAF V600E‑mutant melanoma, further supporting potential efficacy of this combination.
Four partial responses among eight patients (50%) with NRAS‑mutant melanoma were also observed; historically, MEK inhibitors have had limited activity in this subgroup with no partial responses among seven NRAS‑mutant patients in a trametinib phase 1 study and none among ten NRAS‑mutant patients with selumetinib in a larger phase 2 study, whereas binimetinib (MEK162) produced a 21% partial response rate (11% confirmed) and median PFS of 3.7 months with a larger phase 3 study underway, and the present results show superior activity that may provide a future option for this subgroup.
The combination was well tolerated, trametinib could be given at the recommended monotherapy dose, toxicities were generally non‑overlapping, reversible, and consistent with known profiles, the possible treatment‑related fatality occurred amid renal and hepatic injury and overall performance decline, trametinib‑related nephrotoxicity is rarely reported but plausible, and further evaluation in phase 2 is warranted.
In summary, weekly paclitaxel at 80 mg/m2 given with 2.0 mg daily trametinib is tolerable and demonstrates early evidence of efficacy in BRAF wild‑type melanoma, with activity also evident in NRAS‑mutant disease, and a randomised phase 2 trial comparing paclitaxel with or without trametinib is ongoing.
Putting research into context
A PubMed search for reports of MEK inhibitor activity in melanoma published between 1 January 2005 and 18 April 2014 using the terms “MEK inhibitor,” “melanoma,” and “clinical trial,” with a focus on BRAF V600E wild‑type melanoma, identified two trials describing low efficacy of MEK inhibitor monotherapy in patients without a BRAF mutation and a phase 2 trial suggesting better activity when combining a MEK inhibitor with docetaxel, while apart from a single trial, MEK inhibitors alone have not been effective in NRAS‑mutant disease.
Interpretation
The combination of paclitaxel and trametinib is safe with a manageable toxicity profile, activity in an almost exclusively BRAF wild‑type population compares favourably with previous reports, promising activity is also reported in NRAS‑mutated disease, and a phase 2 study is underway to further assess the combination.