Abstract
Periodontitis is an inflammation characterized by alveolar bone resorption caused by an imbalance in bone homeostasis. It is known that autophagy is related to inflammation and bone metabolism. However, whether autophagy inhibitors could be used for periodontitis in animal models remains unknown. We investigated the role of two classical autophagy inhibitors, 3-methyladenine (3-MA) and chloroquine (CQ), on the development of rat experimental periodontitis in terms of the bone loss (micro-CT), the number of inflammatory cells (hematoxylin and eosin staining), and the osteoclastic activity (tartrate-resistant acid phosphatase staining). Expression of autophagy-related genes and nuclear factor kappa B p65 (NF-κB p65) were assessed by immunohistochemistry.
Expression of Beclin-1 and microtubule-associated proteins 1A/1B light chain 3 (LC3) were analyzed by Western blot. To further observe the effect of autophagy inhibitors on osteoclasts (OCs) in vitro, bone marrow–derived mononuclear macrophages were used. Together, these findings indicated that topical administration of 3-MA or CQ reduced the infiltration of inflammatory cells and alveolar bone resorption in experimental periodontitis. Furthermore, 3-MA and CQ may attenuate activation of OCs by autophagy. Therefore, 3-MA and CQ may have prophylactic and therapeutic potential for inflammation and alveolar bone resorption in periodontitis in the future.
INTRODUCTION
Periodontitis is an infectious disease characterized by periodontal inflammation and periodontal attachment loss, subsequently leading to the alveolar bone resorption and loss of teeth . Periodontal inflammation exacerbates the alveolar bone resorption. In addition to merely controlling the inflammation in the periodontium, inhibiting the absorption of alveolar bone has also become a major goal of periodontal therapy.
Bone homeostasis is regulated by a balance between osteoclastogenesis and osteoblastogenesis, which is similar to the relationship between Yin and Yang in Taiji. A disturbance in bone homeostasis leads to increased bone loss and various pathological disorders including periodontitis, osteomyelitis, osteoporosis, and rheumatoid arthritis. Osteoclasts (OCs), which are multinucleated cells of monocyte/macrophage origin, are majorly responsible for mediating bone degradation and are essentially required for bone surface erosions. Taken together, the regulation of osteoclastic activation is the pivotal aim in providing novel treatments for protecting the alveolar bone from excessive absorption in periodontitis.
Autophagy is an evolutionarily highly conserved cellular homeostatic process, which maintains cellular homeostasis depending on the turnover of excess or dysfunctional cellular components following the lysosomal machinery . In view of the pivotal role of autophagy on regulating cellular homeostasis, it may not be surprising that autophagy is involved in the pathogenesis of several diseases including cancer, infections, and cardiovascular diseases. Periodontitis, as an inflammatory disease, is also associated with autophagy.
A higher level of autophagy in gene expression and autophagosome production of periodontal tissues was found in periodontitis patients. In addition, autophagy-related protein ATG7 and Beclin-1, increasing in the OCs of rheumatoid arthritis patients, suggested that increased autophagy in OCs leads to bone absorption in vivo. Defective microtubule-dependent podosome belt formation in OCs has been reported in response to nocodazole, an inhibitor of phosphatidylinositol 3-kinases (PI3K), showing that inhibition of autophagy influences the activity of OCs. Recent evidence has demonstrated that curcumin can inhibit ovariectomy-induced bone loss by reducing expression of RANKL. All of these studies suggest a correlation between autophagy, inflammation, and bone resorption.
Based on the above studies, we noted that the level of autophagy is significantly elevated in pathological states such as inflammation. We hypothesized that regulating autophagy may be a potential therapy for the bone destruction in periodontitis. Hence we chose two conventional autophagy inhibitors, 3-methyladenine (3-MA) and chloroquine (CQ), to observe their effects on the development of periodontitis.
3-MA is an inhibitor of PI3K, a key regulator of autophagy, and it can suppress autophagy by blocking the formation of autophagosome. In addition, 3-MA can inhibit the invasion of highly metastatic cancer cells via blocking the class I and II PI3K. CQ acts as an inactive inhibitor of autophagy, blocking the function of lysosomes and the degradation of phagocytosis, thereby inhibiting the survival of cancer cells and inhibiting tumorigenesis.
In this study, using an experimental periodontitis rat model for the first time, we explored the mechanisms by which autophagy may modulate the osteoclastogenesis to periodontitis-related bone loss.
MATERIALS AND METHODS
Animals and Bacterial Strains
The male Sprague-Dawley rats (weight 200–210 g; Qinglongshan Animal Breeding Field, Nanjing, China) of 7 weeks of age were housed in an air-conditioned room (temperature 21–23 °C, humidity 50–60 °C) with free access to food and water on an auto-cycling 12 h of light/12 h of dark cycle under specific pathogen-free conditions. All rats were equilibrated for 1 week, before periodontitis induction. All experimental procedures described in this study have been approved by the Animal Ethics Committee of Nanjing University and were carried out in accordance with the National Institutes of Health guide for the care and use of laboratory animals.
Porphyromonas gingivalis (P. gingivalis, ATCC 33277) strain was cultured anaerobically in brain heart infusion (BHI) medium at 37 °C with 85% N2, 5% H2, and 10% CO2. The concentrations of P. gingivalis were standardized to an optical density of 1 at 600 nm via a spectrophotometer, which corresponds to 10^9 CFU/mL.
Ligation/P. gingivalis–Induced Experimental Periodontitis Model
Thirty-six Sprague-Dawley rats were randomly allocated into six study groups (n = 6/group): No periodontitis (NP) group, periodontitis (P) group, periodontitis with 3-MA low-dose (P+3-MA-L) group, periodontitis with 3-MA high-dose (P+3-MA-H) group, periodontitis with CQ low-dose (P+CQ-L) group, and periodontitis with CQ high-dose (P+CQ-H) group.
Periodontitis was induced via the ligation method. Briefly, P. gingivalis-soaked 4-0 silk ligatures were ligated firmly and sub-gingivally around the bilateral maxillary second molars on day 0 to facilitate bacterial colonization. Simultaneously, 3-MA and CQ (Sigma-Aldrich, MO, USA) were dissolved in 0.9% NaCl solution respectively. Subsequently, the 90 muL of corresponding concentration of the drug (or 0.9% NaCl solution as control in NP group and P group) was injected at the three palatal gingival sites (mesiopalatal, midpalatal, and distopalatal) of the second molars. The corresponding reagent was injected to the rat, the 10^9 CFU/mL P. gingivalis were inoculated into its sulci by a probe, and the ligature status was checked every 2 days. If ligatures were lost or loose, they were replaced again. Three weeks after treatment, all rats were euthanized for further experimental analyses (Fig. 1).
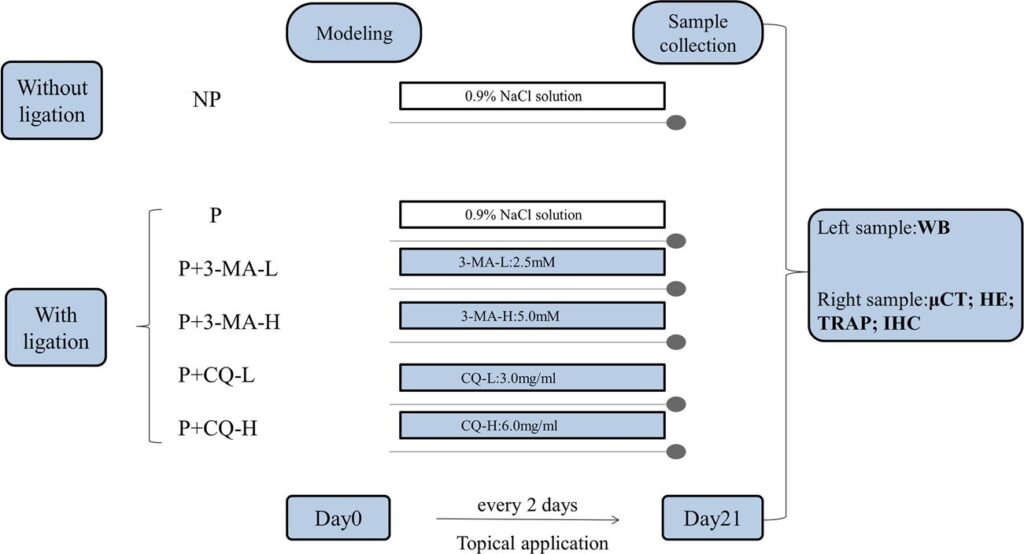
Fig. 1. Experimental design. No periodontitis group (NP), 0.9% NaCl solution application without ligation; periodontitis group (P), 0.9% NaCl solution application with ligation; periodontitis and 3-methyladenine low-dose group (P+3-MA-L), 2.5 mM 3-MA application with ligation; periodontitis and 3-methyladenine high-dose group (P+3-MA-H), 5.0 mM 3-MA application with ligation; periodontitis and chloroquine low-dose group (P+CQ-L), 3.0 mg/mL CQ application with ligation; periodontitis and chloroquine high-dose group (P+CQ-H), 6.0 mg/mL CQ application with ligation. WB, Western blot; muCT, micro-computed tomography; HE, hematoxylin and eosin; TRAP, tartrate-resistant acid phosphatase; IHC, immunohistochemistry.
Micro-computed Tomography Imaging and Bone Resorption Analysis
All right maxillary block biopsies fixed in 4% paraformaldehyde were subjected to be scanned by a micro-computed tomography (muCT) system (Quantum GX; PerkinElmer, USA). Three-dimensional (3D) digitized images of the palatal view were collected for each sample using 3D reconstruction software (Analyze12.0; AnalyzeDirect, Overland Park, KS, 66085, USA), according to the manufacturer’s procedures. Alveolar bone loss was analyzed from 3D mode. Linear measurements were taken from the cemento-enamel junction (CEJ) to the alveolar bone crest (ABC) according to the tomographic method. Briefly, three orthogonal views were shown as cross-hair, the transaxial view (X-Y plane, blue-green line), the coronal view (X-Z plane, blue-red line), and the sagittal view (Z-Y plane, red-green line).
In order to evenly align the anatomical positions and scan axes, these 3 views were reoriented before measurement, including long axis of the second molar was parallel to the Z axis, and long axis of the occlusal surface was parallel to the X axis, and the occlusal surface was parallel to the Y axis. Then, keeping the X and Y axes still, we sequentially measured CEJ-ABC at five sites in the transaxial plane, namely, the two mesiopalatal sites, the one palatal groove site, and the two distopalatal sites by adjusting position of the z axis (Fig. 2b). The mean value of all five sites was determined as experimental data.
Histopathological Evaluation
After scanned, the specimens were decalcified, dehydrated and embedded in paraffin. Serial 4-mum sections in the mesiodistal plane, mounted on adhesive-proof slides, were prepared for hematoxylin and eosin (H&E) staining, tartrate-resistant acid phosphatase (TRAP) histochemical staining, and immunohistochemistry (IHC) staining. For evaluating the histological changes in the periodontal tissues, the inflammatory cells in a fixed area of 10,000 mum^2 at a magnification of times 400 were counted, which could be recognized as neutrophils, lymphocytes, macrophages, or eosinophils based on size, shape, and position. Fibroblasts and squamous epithelial cells in the field of vision were not calculated. The total number of inflammatory cells represented the inflammatory state in the visual field. Three consecutive square fields, involving junctional epithelium (JE), gingival epithelium (GE), and alveolar bone (AB) in the mesial coronal area of the second molar, were selected from each section. Six sections of each group were analyzed, and each measurement was counted by the examiner in a blind manner.
Tartrate-Resistant Acid Phosphatase Histochemistry
For osteoclastic activity analysis, sections were stained using a TRAP (Sigma, St. Louis, MO, USA) according to the manufacturer’s instructions. In brief, after deparaffinized and hydrated, the specimens were incubated with a mixture of TRAP stain for 1 h at 37 °C in the dark.
Fig. 2. Local administration of autophagy inhibitors decreased the CEJ-ABC distance. a Images of the maxillary second molar on palatal showed the alveolar bone loss. b Effects of autophagy inhibitor on alveolar bone loss examined by tomographic method in maxilla secondary molars of rats. Blue line, the X axis; green line, the Y axis; red line, the Z axis; red arrow, the 5 selected measurement sites. The distance between two yellow lines represents each site of the distance from the CEJ to the ABC on palatal. c Differences of alveolar bone levels. P < 0.05; P < 0.01, compared with the periodontitis group. CEJ, cemento-enamel junction; ABC, alveolar bone crest.
Active osteoclasts were defined as TRAP-positive cells, with multiple nuclei containing three or more nuclei along the alveolar bone surface under a light microscope. Osteoclasts around the alveolar bone surface between the first and second molars were counted and analyzed on the basis of the TRAP-stained images. The results from each group were expressed as the mean pm standard deviation (SD).
Immunohistochemistry Analysis
After being deparaffinized, rehydrated, and antigen retrieved, the sample slices were quenched with endogenous peroxidase (3% hydrogen peroxide) and blocked with 3% bovine serum albumin for 30 min. Then, sections were respectively incubated with autophagy-related antibody: Beclin-1 (1:100; Servicebio), p62/SQSTM1 (1:100; Servicebio), LC3B (1:100; Cell Signaling Technology), as well as NF-κB p65 (1:100; Servicebio). Primary antibodies were incubated overnight at 4 °C.
After washing with phosphate-buffered saline (PBS), secondary antibodies were added to the slices for 50 min at room temperature. For visualizing the immunoreactivity, diaminobenzidine (DAB; Servicebio, G1211) from a colorimetric detection kit was used. After being counterstained with hematoxylin (Servicebio, G1004), images were acquired by a digital microscope (3DHISTECH Ltd, Budapest, Hungary). IHC staining images were semi-quantitatively analyzed by Image Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA). Three same areas from the AB to the GE in the second molar mesial in each slice were selected to measure the integrated optical density (IOD) at times 400 magnification. The mean of optical density (MOD), the values of IOD/area, was represented as the final result.
Western Blot
Western blot was performed to evaluate the expression of autophagy-related gene in periodontium. Briefly, after cut into small pieces, tissues including gingiva and connective tissue were lysed in the radio immunoprecipitation assay (RIPA, Beyotime) lysis buffer and phenylmethylsulphonyl fluoride (PMSF, Beyotime). Hereafter, samples were centrifuged at 12,000 rpm at 4 °C for 15 min in three times freezing-melting cycles. The supernatant was collected and protein concentration was initially determined using BCA kit (Beyotime). Fifty micrograms of protein on each sample was separated by 12% SDS-PAGE and transferred onto PVDF membranes (Millipore, Bedford, MA, USA).
After blocking with 5% BSA dissolved in Tris-buffered saline containing 0.5% Tween 20% (TBST) for 1 h at room temperature, the membranes were incubated with the primary antibody overnight at 4 °C. The primary antibodies were purchased from Cell Signaling Technology (Beclin-1, p62, and LC3). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Bioworld, St. Louis, USA) was used as an internal control. After three washes with TBST, the membranes were incubated with corresponding secondary antibody for another 1 h. Immunoreactive bands were visualized using chemiluminescence substrates (Merck Millipore, Darmstadt, Germany) and chemiluminescent imaging system (LAS4000M; GE Healthcare Bio-Sciences Corp., Piscataway, USA).
Cell Culture
To further observe the effects of autophagy inhibitors on OCs, bone marrow–derived mononuclear macrophages (BMMS) were used. BMMS were primarily retrieved from the tibia and femur of 4 to 6-week-old C57BL/6J mice (Animal Model Institute, Nanjing University, China) that were incubated on plates with alpha-minimal essential medium (alpha-MEM, Gibco, Thermo Fisher Scientific, Waltham, USA) containing 15% foetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (HyClone, Logan, USA) at 37 °C in 5% CO2 for 18 h. Then cells in the supernatant were centrifuged and resuspended in alpha-MEM, and then maintained in alpha-MEM containing 30 ng/mL recombinant murine macrophage colony–stimulating factor (M-CSF, PeproTech, USA) for 7 days.
Pre-osteoclasts (pre-OCs) were generated by incubation with 30 ng/mL M-CSF and 50 ng/mL RANKL. After 4 days of incubation, the cells were pre-treated with 40 muM 3-MA and 4 muM CQ 1 h before stimulation with 500 ng/mL Escherichia coli–derived lipopolysaccharides (LPS; Sigma-Aldrich) and the culture medium was changed with the same new medium on day 2. After 3 days of incubation, cells were fixed in 4% PBS paraformaldehyde for 20 min and stained with TRAP and TRAP-positive cells per well were counted.
Statistical Analysis
All data was expressed as mean pm SD. The difference between two groups was conducted using an unpaired t test and the difference among three or more groups was analyzed using a one-way analysis of variance in GraphPad Prism 6.0 software (GraphPad Software, USA). A two-tailed p < 0.05 was considered to indicate statistical significance.
RESULTS
Local Administration of Autophagy Inhibitors Decreased the CEJ-ABC Distance and Reduced the Number of Osteoclasts
The overall morphology as a reconstructed 3D model and the linear bone loss on palatal are shown in Fig. 2a, in which the P group presents the highest level of alveolar bone loss. Measurement of the CEJ-ABC distance is displayed in Fig. 2b. As it is shown, the CEJ-ABC distances were decreased by high-dose CQ in Fig. 2c. Analogous CEJ-ABC distance reduction occurred in the P+3-MA-L, P+3-MA-H, and P+CQ-L groups, although not statistically significant.
In view of the inhibition of bone loss, we then analyzed the numbers of OCs in areas related with the site of injections in the gingival tissues since OCs are primarily responsible for bone resorption. Mean osteoclast counts of the groups were presented in Fig. 3a. The number of the OCs increased significantly in the P group in accordance with the amount of bone loss. Expressions of TRAP-positive cells were inhibited in the P+3-MA-L, P+3-MA-H, P+CQ-L, and P+CQ-H groups (Fig. 3b).
Fig. 3. Effect of autophagy inhibitors on the number of osteoclasts. Supplementation with 3-MA and CQ prevents the formation of active osteoclasts in experimental periodontitis rats. a Osteoclasts were stained red by tartrate-resistant acid for phosphatase staining. Scale bar, 200 mum (up); 100 mum (down). b Statistical analysis for the number of osteoclasts (P < 0.001, compared with the periodontitis group).
Inflammation Was Attenuated by Local Administration of Autophagy Inhibitors
H&E staining analysis exhibited ordinary structure including the gingivae, periodontal ligament, cementum, and alveolar bone in the NP group. In the P group, areas between the first and the second molars exhibited extensive damage including the intense inflammatory cell infiltration; alveolar bone loss; the gingival-combined epithelium shifted to the root, forming a periodontal pocket. Periodontal damages in the periodontitis groups with autophagy inhibitors were apparently retrieved (Fig. 4a). As it is shown in Fig. 4d, the number of inflammatory cells in the junctional epithelium was increased in the P group, and decreased significantly in P+3-MA-L, P+3-MA-H, P+CQ-L, and P+CQ-H groups.
NF-kB was closely related to the expression of various pro-inflammatory mediators; thus, NF-κB p65 was stained by immunohistochemistry to evaluate the level of inflammation in the periodontal tissues corresponding to the site of injections (Fig. 4b). NF-κB p65 was significantly escalated in periodontitis groups, and was clearly attenuated by the local administration of 3-MA or CQ, supporting anti-inflammatory effects of autophagy inhibitors (Fig. 4e).
Fig. 4. Local administration of autophagy inhibitors attenuated the number of inflammatory cells and the expression of NF-κB p65. a Image of hematoxylin and eosin staining, presented the status of periodontium region in the maxillary second molar. Notable inflammatory cell infiltration could be seen in the periodontitis group. Connective tissue destruction, the irregularly arranged fibroblast, and alveolar bone resorption could be found in the interdental area. Scale bar, 200 mum (up); 50 mum (down).
Expression of NF-κB p65 in the periodontium. Scale bar, 200 mum (up); 50 mum (down). c Schematic illustration of the target regions of inflammatory cells of rat periodontium. The three successive square views (100 times 100 mum) from the junctional epithelium (JE) to the gingival epithelium (GE) and the alveolar bone (AB) demonstrate the target areas that were used to count inflammatory cells. d Statistical analysis for the number of inflammatory cells in the periodontium. e The semi-quantitative analysis of the NF-κB p65 expression in periodontal tissues. (P < 0.01; P < 0.0001, compared with the periodontitis group). At least three slides per each experiment were observed: alveolar bone (AB) and dentine (De).
Local Administration with Autophagy Inhibitors Decreased the Level of Autophagy in Periodontal Tissue
IHC staining exhibited the expression of Beclin-1, p62, and LC3B in periodontal tissue (Fig. 5a), and Western blotting shows the expression of LC3 and Beclin-1 (Fig. 5d). The expression level of Beclin-1 (Fig. 5b and f), LC3B (Fig. 5d), and LC3-II (Fig. 5g) was significantly elevated in the P group; however, the level of p62 (Fig. 5c) remained decreased. The results suggested the autophagy was significantly induced in the periodontitis group. Opposite results, decreasing Beclin-1 and LC3-II and increasing p62, were presented by application with 3-MA or CQ, indicating a marked autophagy suppression.
Fig. 5. Level of autophagy was decreased by local administration with autophagy inhibitors. a Expression of Beclin-1, p62, and LC3B. In the group with no periodontitis, Beclin-1, p62, and LC3B were moderately expressed. In inflamed tissues from the periodontitis group, the very strong immunoreactivity of Beclin-1 and LC3B could be found in most fibroblast cells, bone surfaces, and root surfaces, and barely no positive staining of p62 could be found. Scale bar, 200 mum (up); 50 mum (down). b–d The corresponding semi-quantitative analysis of the Beclin-1, p62, and LC3B expression in periodontal tissues. e–g Expression of Beclin-1 and LC3-II as determined in the gingival tissue of the rats by Western blot analysis. Beclin-1 and LC3-II expression was downregulated with 3-MA or CQ, as assessed via immunohistochemistry. (P < 0.05; P < 0.01; P < 0.001; P < 0.0001, compared with the periodontitis group).
Autophagy Inhibitors Attenuated LPS-promoted Osteoclastogenesis In Vitro
After 4-day induction, M-CSF and RANKL stimulated the osteoclastogenesis of BMMs and LPS significantly increased the number of mature OCs (Fig. 6a). To explore whether the activation of autophagy has an effect on the osteoclastogenesis, we employed 3-MA and CQ in LPS-treated BMMS. The increased osteoclastogenesis caused by LPS was evidently restrained by 3-MA or
Fig. 6. Effect of autophagy inhibitors 3-MA and CQ on LPS-stimulated mouse bone marrow–derived macrophages to form osteoclasts. LPS increased the number of mature OCs significantly (a). Increased osteoclastogenesis caused by LPS was evidently restrained by 3-MA or CQ (b).
DISCUSSION
Periodontitis is a common inflammatory disease, where the chronic inflammatory irritation can lead to absorption of alveolar bone and subsequent tooth loss. Numerous studies have suggested that inflammation and autophagy regulation occur in response to the stimulation of P. gingivalis, the main pathogen of periodontitis, and increasing researches have discussed the effect of autophagy on osteoclasts in vitro, and the effect of autophagy on the bone destruction in vivo. Despite the unclear mechanism, inflammatory cell infiltration induced by bacterial plaque biofilm and bone resorption induced by osteoclasts have been shown to be inextricably linked to autophagy.
Autophagy plays a crucial role in physiological and pathological processes. In previous studies, an increasing level of autophagy could be found in patients with periodontitis compared with no periodontitis. Based on the importance of homeostasis of autophagy to pathological conditions, we hypothesize that autophagy modulation, mainly inhibiting an excessive autophagy induced by inflammation, may be a potential prophylactic and therapeutics in inflammatory bone resorption in periodontitis.
Autophagy can be interfered with by pharmacological and genetic means. As the initial discussion, we had chosen pharmacological methods, which are relatively easy to perform. We speculated that two chemical autophagy inhibitors, CQ and 3-MA, may inhibit alveolar bone resorption via inhibition of autophagy on the course of periodontitis. Under inflammation, 3-MA abolishes the early stage of autophagy by its prohibitive effect on class III PI3K activity. CQ, which is used as an antimalarial and antirheumatoid drug, inhibits the late states of autophagy by blocking the fusion of autophagosomes with lysosomes. We selected the concentrations of 3-MA and CQ based on previous reports.
In our study, muCT analysis revealed that treatment with P. gingivalis–adhered ligatures caused alveolar bone loss. Compared with a high dose of CQ, low dose of CQ and high and low doses of 3-MA did not provide protection in bone resorption by muCT significantly. The histological analyses showed both CQ and 3-MA inhibited alveolar bone resorption and periodontal destruction. This may be the statistical result caused by the fact that the doses of 3-MA used for local treatment were not enough and the molecular weight of 3-MA (149.15), being lower than that of CQ (515.86), may cause a different distribution mode and function efficiency for the two autophagy inhibitors.
To verify the effectiveness of local injection of 3-MA or CQ, we analyzed three autophagy-related genes (Beclin-1, LC3, and p62/SQSTM1) by Western blot analyses and IHC staining. In our study, we found that the ratio of LC3-II and expression of Beclin-1 and LC3B, the essential autophagy proteins, were significantly enhanced in the P group but was diminished upon 3-MA/CQ treatment, while p62/SQSTM1 had an opposite tendency. From these results, excessive autophagy level could be found in the periodontitis induced by ligation and local administration of 3-MA/CQ did inhibit autophagy in gingival tissues.
Autophagy appears to regulate bone loss primarily by its effects on OCs activation and differentiation. We detected OCs formation by TRAP staining in vivo and in vitro. The in vitro results showed the 3-MA/CQ group showed lower number of TRAP-positive multinucleated cells compared with the LPS group, which were consistent with the in vivo findings and were in parallel with the previous report indicating that 3-MA decreased the LPS-induced OCs formation. Our previous study suggests that inhibition of bone loss by 3-MA/CQ may depend on direct impairment of OCs activation.
These data were consistent with a previous in vitro study which showed 3-MA negatively regulated osteoclastogenesis. Meanwhile, an interesting phenomenon caught our attention, there was a little more osteoclasts in the 3-MA group than in the 3-MA+LPS group (Fig. 6b), in spite of no statistically difference between these two groups (p = 0.066 > 0.05). Wu et al. had found that 3-MA could promote autophagy flux when treated under nutrient-rich conditions, whereas it still suppressed starvation-induced autophagy, suggesting 3-MA has a dual effect on modulation of autophagy. Therefore, we speculated that 3-MA may have different effect on osteoclasts with or without LPS stimulation, but further study is still needed to verify this conjecture and to investigate the underlying specific mechanism and more caution should be exercised in the application of 3-MA in autophagy study.
In addition, autophagy is also of great importance for controlling inflammatory responses. To explore whether 3-MA/CQ has an anti-inflammatory effect on periodontitis, we examined the alteration of inflammatory cells and inflammatory response–related protein, NF-κB p65, which is considered a crucial component of the inflammatory response. Previous studies have indicated a regulatory crosstalk between autophagy and NF-κB signalling cascade. In the present study, we first explored in experimental periodontitis whether 3-MA/CQ treatment inhibited the phosphorylation of NF-κB p65, suggesting that NF-κB p65 acts downstream of the class III PI3K in the process of autophagy in OCs. Both doses of 3-MA or CQ prevented the inflammatory cells infiltrating toward the junctional epithelium, which indicated autophagy may play a role in inflammation in periodontitis.
In conclusion, these findings from our study suggest that autophagy plays vital roles in the inflammation and the bone resorption process in periodontitis. Pharmacological inhibition of autophagy, via 3-MA or CQ, may reduce inflammation, osteoclastogenesis, and bone resorption in experimental periodontitis with excessive autophagy level. Notably, pharmacological interference with autophagy is a complex process and what is the “normal” range of autophagy activity remains not fully elucidated. Thus, maintaining homeostasis of autophagy could be promising candidates for novel drugs for periodontal disease. Our further research will explore this mechanism in order to understanding the balance between autophagy and inflammation in periodontitis.